Limited Uptake of Biosimilars Despite Potential Cost Savings: Patent Expiration and Value-Based Contracting to Drive Market Changes
Summary: Biosimilars are drugs that reference a biologic and are concentrated in high-cost specialty drug markets such as oncology. Despite potential for cost savings in Medicare and other markets, uptake of biosimilars in the US has historically been low because of rebate incentives for high-cost drugs, limited patient and prescriber awareness, ASP-based reimbursement, formulary placement, patent thickets, and state substitution laws. The upcoming patent expiration for the blockbuster drug Humira and different approaches to reimbursement represent an opportunity for US stakeholders to capitalize on the potential for healthcare system savings and reach utilization levels similar to those in Europe. In the next 2-3 years, increased familiarity with biosimilars as well as the first interchangeable products reaching the market will likely accelerate the adoption of these less-costly alternatives in immunology and oncology, aided by alternative payment models.
Biosimilars are biologic drugs approved through an abbreviated FDA pathway introduced during the passage of the ACA, when a product sponsor demonstrates through clinical trials that they have no clinically meaningful differences with the originator products. In May 2022, 35 biosimilars have been approved by the FDA, while many others have not yet launched because of litigation and patent settlements. Although this pathway has been available since 2010, the first biosimilar was first approved in the US in 2015. Additionally, a biosimilar can be designated as interchangeable if a manufacturer demonstrates that there are no differences between the two products. Notably, 2 products have now been designated as interchangeable which first happened to the insulin product Semglee in July 2021, and for the immunology Humira reference product Cyltezo in October.
Due to the existing rebate system, pharmaceutical benefit manager (PBM) practices, and provider reimbursement payments, many stakeholders are not incentivized to prescribe biosimilars. For example, in Medicare Part D, drug manufacturers may offer prescription drug plan sponsors higher rebates than their biosimilar counterparts for better formulary tier placement. State Medicaid programs are guaranteed statutory rebates for drugs approved by the FDA but can negotiate supplemental rebates and use drug utilization management techniques such as prior authorization to promote lower-cost alternatives. For providers, barriers to prescribing include a lack of awareness of different prescribing options, concerns with switching patients to a new treatment regimen that may be less effective, and reimbursement based on average sales price add-on being lower for biosimilars than brand-name drugs. Additionally, the terms used by the FDA (e.g., interchangeable) may lead different stakeholders to assume that biosimilars are inferior or not as effective than the reference product and create additional uncertainty.
A recent report from the HHS Office of the Inspector General (OIG) highlighted that increased biosimilar use in Part D could drive down overall drug costs but face challenges in prescribing, formulary placement, and utilization data. The report found that the brand-name reference products were still prescribed as 5 times as frequently as biosimilars in 2019, even though biosimilars could reduce beneficiaries out-of-pocket spending by 12% if they were prescribed at higher rates. There are 8 approved biosimilars in the Part D market that treat autoimmune diseases, neutropenia, and chronic kidney disease and are self-administered. Zarxio has had the most success in achieving higher market share since its approval of the first biosimilar in the US market in 2015. Zarxio and other filgrastim biosimilars, which are used to treat neutropenia by increasing chemotherapy patients’ white blood cell counts to prevent infections, have achieved 62% of market share compared to the originator product Neupogen. Meanwhile, market share in Part D for other classes of biosimilars in 2019 have only reached 7% to 16% of market share. If other classes of biosimilar drugs achieved utilization rates of 60% on par with filgrastim biosimilars, OIG estimates that Part D spending in 2019 could have been lowered by $84 million dollars, and direct beneficiary out-of-pocket spending could have been reduced by up to $1.8 million. These cost savings are especially pronounced during the initial coverage phase when there is higher beneficiary cost-sharing (compared to the catastrophic phase) and could be even higher enhanced if biosimilar usage reaches the same rate as generic drugs (~90%). This is partly because Medicare formularies may not cover the biosimilar or place a biosimilar competitor in the same tier as their reference product (88- 97% of formularies by drug class). Plans often have the same prior authorization and step therapy requirements for biologics and their biosimilar counterparts (85-95% by drug class), with Zarxio being a notable outlier.
Although new biosimilar products are coming to the market in 2023 for Humira, a multi-billion-dollar blockbuster drug prescribed for arthritis, psoriasis, and Crohn’s disease, plan formulary coverage and utilization management practices may not fully incentivize biosimilar use in Medicare Part D formularies based on existing trends and structural barriers in the US. Even though this upcoming patent expiration is a vital opportunity for Medicare to reduce Part D spending with 7 Humira biosimilars coming to market next year and 1 of them designated as interchangeable, significant rebate offerings may negate the potential market share shifts and lower costs.
CMS has taken some steps to facilitate biosimilar uptake. The MA and Part D 2021 Final Rule created an option for sponsors to create formularies with a preferred and non-preferred specialty tier of drugs to differentiate between high-cost biologic drugs and biosimilar counterparts. However, this change may be adopted slowly due to the additional complexity and administrative costs required. CMS also promotes biosimilar use through the CMS Innovation Center (CMMI) value-based care models that promote the use of lower total costs of care. CMMI’s strategic refresh released in August of 2021 under the Biden Administration promised to increase biosimilar use and reduce costs through models that incentivize shared savings and bundled payments. Although details were limited, CMMI pointed to the Part D Senior Savings Model as a demonstration that could be expanded to include biosimilars (the existing model launched in 2022 offers limited insulin copays during the first 3 phases of Part D coverage as a supplemental benefit).
Even though there is widespread anticipation for the next round of alternative payment models and demonstrations under the Biden Administration, CMMI has not yet announced any new models that target drug pricing specifically or that will replace the Oncology Care Model (OCM), despite its success in incentivizing Part B biosimilar use through bundled payments. One study found that biosimilar use has helped the success of the OCM as payments for breast cancer drug Herceptin were 13-35% lower than the originator and 21-27% lower for another common oncology drug, Avastin. The model also helped accelerate the use of filgrastim biosimilars and save providers participating in the OCM money in breast cancer, lymphoma, and colon cancer drug costs (14%, 8%, and 14% respectively) due to high costs of infusion drugs used in oncology care.
CMS provided biologics with their own HCPCS billing codes in 2018 for documentation and tracking, while providers’ add-on payment for administering a biosimilar is based on the average sales price (ASP). Since biosimilar reimbursement rates are lower than the originator products, providers are not strongly incentivized to switch to a lower cost alternative even though prescribing a brand-name brand results in higher patient out-of-pocket costs for patients based on a set coinsurance rate. Different policy recommendations for reforming the current ASP +6% reimbursement policy to promote biosimilar prescribing include increasing the reimbursement for biosimilars above the originator (e.g., ASP+8%), creating shared savings programs for physicians to reap the rewards of biosimilars, implementing a “least-costly alternative” payment rate, or a using blended reimbursement rate for both products. For example, a blended reimbursement rate of the volume-weighted average of both products would create downward pressure on the price of all drugs in each class and increase the financial risk of prescribing a more costly alternative if there is no clinical difference. However, any changes could greatly disrupt biologics markets that have much higher development costs than generics and would require legislative action or a CMMI model.
Federal legislative efforts so far have focused on increasing patient and provider education through the “Advancing Education on Biosimilars Act” that was enacted in April 2021. It directs federal agencies to create education programs for providers and other educational materials for the public that build upon what is provided by the FDA. Many FDA documents on provider educational materials, pre-approval safety review, and biosimilar development have recently been updated to deemphasize the need for certain additional safety studies, such as animal studies. At the same time, “false and misleading” statements about the comparative effectiveness of biosimilars by competitors have increased confusion and led the FDA and FTC to release a joint press release on prevention efforts in 2020. Separately, federal legislation has proposed to add a biosimilar star rating measure for Medicare Advantage plans so that consumers can identify plans that have better biosimilar access through formularies, cost-sharing, and utilization management.
Medicaid has limited evidence on the prevalence of biosimilar uptake, which is largely determined by physician prescribing because of state pharmacy requirements. However, some states choose to control the management of the pharmacy benefit for Medicaid enrollees and are more closely involved with coverage decisions, while many states contract these services out to a PBM. However, recent research in New York, which carves out their pharmacy benefits in Medicaid have shown that initiatives in provider education and outreach by pharmacy teams can help to drastically increase the use of biosimilars over originator drugs.
Other policy roadmaps for increasing competition in the biosimilar drug market include the Drug Pricing Plan published by the HHS ASPE office in 2021 which recommended that policymakers: re-evaluate the exclusivity period of biologic patents, streamline the regulatory approval process and clinical trial data needed by the FDA, transition to value-based or indication-based pricing, and increase the use of outcomes-based contracting. Other more complex solutions would have to address the need for patent thicket reform that many manufacturers use to solidify market dominance and deter competitors, which has prevented many biosimilars from coming to market because of “pay-for-delay” agreements. Despite this, many drug patents are set to expire in the next few years and could more than double the number of drugs with biosimilar competition by 2026. While biosimilars coming to market in 2023 are spread between many different therapeutic areas, the next 5 years will see major patent expirations in oncology, immunology, diabetes, and eye treatments (i.e., wet age-related macular degeneration).
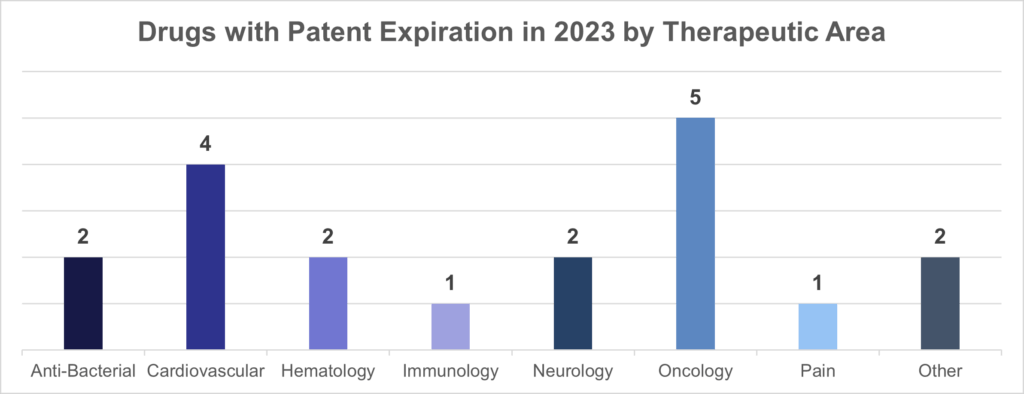
Source: GreyB, 107 Drug Patents that are Expiring between 2023 to 2025. Available here.
At the state level, all 50 states now have laws permitting biosimilar substitution for commercial insurance coverage by a pharmacist when a drug is interchangeable, resulting in more restrictions for biosimilars compared to laws for generics. State laws are highly protective of the physician/patient prescribing relationship and require that a product must be interchangeable due to widespread concerns that patients would not respond as well to another drug forced on them by insurers to lower overall costs. Typical state laws require that patients and physicians must be informed of the switch and providers are allowed to prevent a substitution if deemed medically necessary. Some laws may also require: a record of any change to be kept by the pharmacist, a list of interchangeable products be maintained by the state pharmacy board, or for the pharmacist to notify the patient of lower cost alternatives. These laws have likely decreased biosimilar adoption in the commercial market by imposing strict requirements on pharmacists due to concerns related to efficacy and immunogenicity (a lowered immune response when switching patients with autoimmune diseases between biologic treatments). However, a growing body of research has shown comparable differences in efficacy and safety but demonstrate the need for continued real-world evidence generation.
Comparatively, the European market has been highly successful at increasing biosimilar uptake. According to a recent study, the EU has approved a much larger number of biosimilars than the US (84 versus 35), achieved 30-80% price discounts across different countries, and may mandate price reductions for new biosimilar market entries. The EU also rarely awards process patents that can be used to extend the period of exclusivity, which is common in the US and can increase the length of a patent from 12 years to more than 20 years for a drug like Humira with multiple add-ons. The European Medicines Agency (EMA) that approves new drug applications also created regulatory guidance documents that were specific to drugs in the same biologic class (e.g., insulins), which the FDA did not do in order to protect manufacturer’s intellectual property rights. On the policy side, some countries like Norway make biosimilar substitution automatic, in stark contrast with the majority of US state-level pharmacy substitution laws. For these reasons, in addition to other country-specific factors like Denmark’s system that has central planning of drug recommendations and awards national drug contracts to just one manufacturer, biosimilars have seen much higher rates of market penetration in the EU compared to the US.
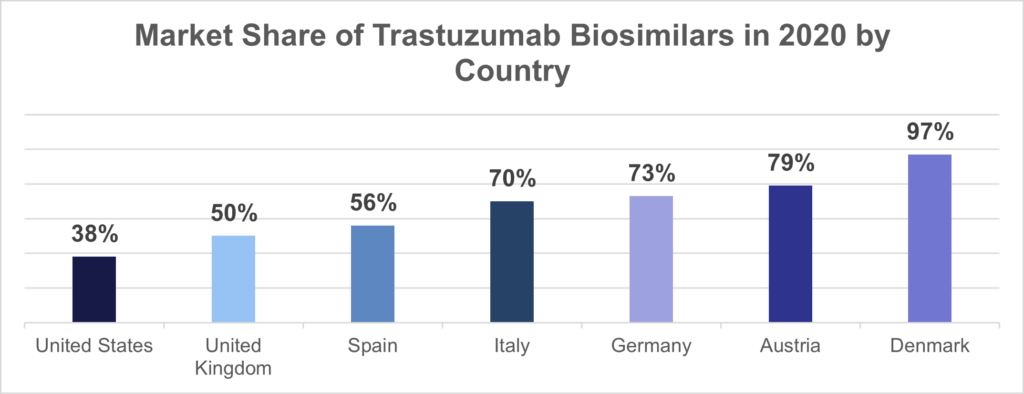
Sources: Azuz, S., Newton, M., Bartels, D. et al. “Uptake of Biosimilar Trastuzumab in Denmark Compared with Other European Countries.” 2021. Available here. IQVIA: Biosimilars in the United States 2020-2024. Available here.
Future Outlook for Biosimilar Uptake in the US
Despite limiting factors, the growth of the biosimilar market in the US is poised to accelerate over the next 5-10 years as a number of blockbuster drugs reach the end of their patent exclusivity period and biosimilar use becomes more widely accepted by stakeholders. Policymakers can accelerate this shift through CMMI demonstrations that build upon the successes of the OCM and continue to make manufacturer and PBM contracting of rebates more transparent, while private and public payers can continue outreach in specific therapeutic areas with the potential for greater use of less costly alternatives. More drugs being designated as interchangeable by the FDA can also increase uptake of in the US by allowing pharmacists to substitute brand-name drugs for biosimilars.
Provider practices can take initiative to directly engage with commercial payers to establish value-based care contracts that emphasize lower overall oncology care costs using biosimilars. Clinicians can also discuss the tradeoffs directly with patients through direct engagement and shared decision-making. There is also growing momentum for employer-sponsored plans to more fully embrace the cost-savings of biosimilars through provider education, prescribing incentives, value-based contracts, and formulary design. Medicaid plans who manage their pharmacy benefit can also influence prescribing through education and outreach to doctors to spur uptake of new biosimilars coming to market.
While stakeholders wait for new CMMI models that could increase biosimilar uptake in Medicare Part B and D, oncologists, nephrologists, rheumatologists, and other specialists should stay up to date on the newest biosimilar products reaching the market and the emerging evidence on their comparative efficacy. Going forward, the effects of biosimilar prescribing will be felt by patients that rely on their doctor’s advice for the best care and bear the differences in costs between biosimilars and brand-name biologics.